| |
 总生存期达26.5个月!肺癌靶向治疗耐
作者:Tony
EGFR-TKI耐药后的治疗选择一直是困扰肺癌患者的重要难题。如今,我们对EGF
总生存期达26.5个月!肺癌靶向治疗耐
作者:Tony
EGFR-TKI耐药后的治疗选择一直是困扰肺癌患者的重要难题。如今,我们对EGF
 肺动脉阻塞,胸闷气短,下一步如何处
请教各位有经验的友友们,目前这种情况如何处理?现在气短的厉害,已无法正常行走,走
肺动脉阻塞,胸闷气短,下一步如何处
请教各位有经验的友友们,目前这种情况如何处理?现在气短的厉害,已无法正常行走,走
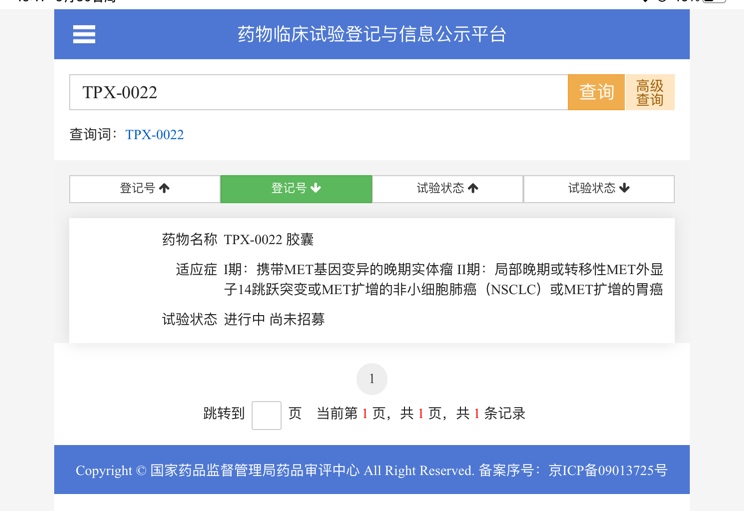
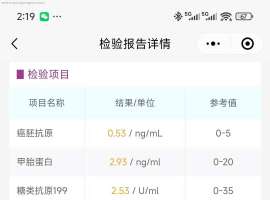 求助:1a1实体型低分化alk突变复发概
背景:35岁男性,无吸烟史,有熬夜习惯,检查前曾参与新房装修。24年7月底体检第一次
求助:1a1实体型低分化alk突变复发概
背景:35岁男性,无吸烟史,有熬夜习惯,检查前曾参与新房装修。24年7月底体检第一次
 二十三个月跋涉:在生命的悬崖边,为
二十三个月。
放在漫长的人生里,不过是弹指一挥间。但对我而言,这几百个日夜,却是
二十三个月跋涉:在生命的悬崖边,为
二十三个月。
放在漫长的人生里,不过是弹指一挥间。但对我而言,这几百个日夜,却是
 破局疑难病例:间质性肺炎风险、脑膜
整理:雨过天晴审核:张蕻梅、钱哲、鹰版在应对肺癌这一复杂疾病时,多学科综合诊疗
破局疑难病例:间质性肺炎风险、脑膜
整理:雨过天晴审核:张蕻梅、钱哲、鹰版在应对肺癌这一复杂疾病时,多学科综合诊疗





 显身卡
显身卡